Epigenetic Modifications in Cancer Etiology, Diagnosis and Therapy
-
Yahaya Tajudeen


Department of Biological Sciences, Federal University Birnin Kebbi, PMB 1157, Kebbi State, Nigeria
Emmanuel JohnDepartment of Biochemistry and Molecular Biology, Federal University Birnin Kebbi, Kebbi State, Nigeria
Asanka Sudeshini HewageInstitute of Biochemistry, Molecular Biology and Biotechnology, University of Colombo, Sri Lanka
Umar Usman LimanInstitute of Biochemistry, Molecular Biology and Biotechnology, University of Colombo, Sri Lanka
Omede CalebDepartment of Biochemistry, Kogi State University, Nigeria
| Received 06 Dec, 2024 |
Accepted 15 Feb, 2025 |
Published 30 Jun, 2025 |
Epigenetic modifications are implicated in the etiologies of various diseases, with cancer being a prominent example. Cancer, a debilitating disease, stands to benefit significantly from advances in the field of epigenetics. Unfortunately, epigenetics has not received sufficient attention and has not been integrated into mainstream healthcare. This study aims to raise public awareness about the link between epigenetics and disease etiologies, with a particular focus on cancer therapeutics. Relevant information was retrieved from reputable academic databases, including Embase, PubMed, Scopus and SpringerLink. The results indicate that epigenetic modifications, mainly noncoding RNA silencing, DNA methylation and histone modification, are essential for growth and development. However, aberrant epigenetic modifications or those exceeding normal levels can lead to diseases. Specifically, abnormal epigenetic changes in tumor suppressor genes (e.g., TP53, RB1, NF1, NF2, CDKN2A, WT1, BRCA1, BRCA2, PARP-1, VHL, APC, PTEN, PTCH1 and CDH1), oncogenes (e.g., RAS, EGFR, EML4AK, LINE-1 and SAT2) or DNA repair genes (e.g., MSH2, MSH6, MLH1, PMS1 and PMS2) can result in cancer. Furthermore, some epigenetic changes are reversible, suggesting that therapeutics targeting these changes in predisposed individuals could be more effective. Epigenetic tests such as Epicup®, Cologuard® and EpiproColon®, along with epigenetic drugs like Azacitidine, Belinostat and Tubacin, have been developed for cancer treatment. However, these drugs face challenges related to poor pharmacokinetics and safety due to a lack of specificity and off-target effects. These issues are currently being addressed with epigenomic therapies. Health professionals are encouraged to target epigenetic changes in predisposed individuals to achieve better outcomes.
| Copyright © 2025 Tajudeen et al. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
Epigenetics is an evolving field of biology focused on disruptions in gene expression that are not attributed to changes in the DNA sequence itself but rather to modifications in the chemical tags on the DNA and associated proteins1. These alterations in gene expression are heritable and influenced by environmental factors2-4. Epigenetic changes can be classified as direct or indirect. Direct changes refer to alterations occurring during an individual's lifetime as a result of direct interactions with their environment5. Indirect changes include modifications that occur in the womb due to events during gestation, as well as changes affecting an individual’s ancestors (such as parents or grandparents), originating from events that took place long before conception5. Epigenetic mechanisms are crucial for normal growth and development, homeostasis and cell and tissue differentiation6. They also play a critical role in regulating pluripotency genes, which become inactivated during differentiation6. In multicellular organisms, epigenetic changes allow different adult cells to express the specific genes required for their unique functions and pass this information on to daughter cells7. Genome-wide patterns of DNA and histone modifications are established during early development and remain stable across numerous cell divisions8. Epigenetics also provides a means for organisms to integrate and react to environmental cues9. Epigenetic modifications influence human appearance, behavior, stress responses, disease susceptibility and even longevity, helping to shape an individual’s phenotype10. Cellular processes associated with epigenetic mechanisms encompass a variety of phenomena such as bookmarking, paramutation, genomic imprinting, gene silencing, position effects, X chromosome inactivation, variability in disorders or phenotypic outcomes, maternal inheritance patterns, carcinogenesis, reprogramming, teratogenic effects, heterochromatin formation, cloning and regulation of histone modifications7.
Incorrect epigenetic modifications or those beyond what is required for normal processes can lead to birth defects, childhood diseases or the emergence of disease symptoms later in life7. Abnormal epigenetic changes have been linked to the development of various diseases, including type 1 diabetes11, type 2 diabetes12, hepatocellular carcinoma13, breast cancer14 and reproductive anomalies15, among others. Fortunately, some epigenetic modifications are reversible, encouraging many researchers to focus on developing epigenetic therapies7. Therefore, targeting the epigenome to treat and prevent diseases represents a promising therapeutic strategy, particularly for debilitating diseases10,16.
Cancer is among the top debilitating diseases that may benefit from improved understanding and breakthroughs in the field of epigenetics. Cancer is the leading cause of death globally, with mortality rates surpassing those of HIV/AIDS, tuberculosis and malaria combined17. It ranks as the second leading cause of death in developed countries and is one of the top three causes of death for adults in developing countries17. In 2022, an estimated 20 million new cancer cases were reported worldwide, along with 9.7 million deaths attributed to cancer18. Currently, over 53.5 million individuals are living with the disease18. Approximately one in five people will develop cancer at some point in their lives, with the mortality rate being about one in nine for men and one in twelve for women18. In Nigeria, annual statistics indicate around 72,000 cancer-related deaths and approximately 102,000 newly diagnosed cases17.
Cancer comprises a range of diseases characterized by abnormal cell growth that can invade or spread to other areas of the body19. Conventional treatment approaches for cancer include surgery, chemotherapy and radiotherapy. Recent advancements in treatment options encompass stem cell therapy, targeted therapy, ablation therapy, nanoparticles, natural antioxidants, radionics, chemodynamic therapy, sonodynamic therapy and ferroptosis-based therapy20. However, these treatments face several challenges, including their suitability being limited to certain molecular subtypes of cancer, high costs, limited availability and the development of resistance in tumors21. Several research initiatives on cancer therapies are ongoing to find better or complementary strategies, with some focusing on epigenetic regulation. It has been established that various stages of tumor progression-such as tumorigenesis, promotion, progression and recurrence-are associated with epigenetic changes, some of which may be reversed through the use of epigenetic drugs22. To this end, some cancer epigenetic drugs, such as I-BET151, decitabine and azacitidine, have shown promise23,24. Nevertheless, awareness of the role of epigenetic mechanisms in cancer pathogenesis and treatment remains limited. This review aimed to clarify and highlight the significance of epigenetic modifications in both the causes and treatments of cancer.
EPIGENETIC MECHANISMS
Epigenetic mechanisms provide an extra level of control within cells, influencing which genes are activated or silenced8. This regulatory process differs among tissues and plays a vital role in cell differentiation8. Additionally, variations in gene expression caused by epigenetic changes are key to the specialized functions of different cell types8. Unlike genetic inheritance, epigenetic marks are shaped by factors such as lifestyle, environment and nutritional status10. There are three primary mechanisms through which epigenetic regulations occur: Noncoding RNA (ncRNA) silencing, histone modification/chromatin remodeling and DNA methylation25-27. These mechanisms can individually influence gene expression or synergistically regulate it15.
LINK BETWEEN EPIGENETIC REPROGRAMMING AND CANCER
The connection between epigenetics and cancer has been intensely studied over the past two decades, generating substantial clinical data that attest to the involvement of epigenetics in disease modulation and therapeutics28-30. The formation of aberrant epigenetic reconfigurations is a hallmark of cancer cells. Aberrant epigenetic modifications in DNA repair genes, tumor suppressor genes (TSGs) or oncogenes, can cause cancer31. A TSG, also known as an oncogene suppressor or anti-oncogene, regulates a cell during division and replication, while oncogenes are mutated genes capable of causing cancer. The DNA repair genes repair damaged DNA. Hypermethylation of TSGs and hypomethylation of oncogenes and DNA repair genes can lead to cancer32,33. Abnormal histone posttranslational modifications can alter the expression of these genes and cause cancer27,34. Moreover, changes in ncRNA expression play crucial roles in human malignancies35. The ncRNAs can act as oncogenes or suppressors, regulating cancer initiation and progression36. Many miRNAs, in particular, function abnormally in cancer cells, contributing to cancer initiation. For instance, miR-126 overexpression downregulates EGFL7 and p53, causing hepatocellular carcinoma and breast colorectal cancers, respectively36,37. The miR-155 is highly expressed in many cancers, such as breast, colon, lung, liver and gastric cancers38. Furthermore, MiR-215 is overexpressed in glioblastoma in people undergoing hypoxia39. Some miRNAs, regarded as tumor suppressors, such as miR-34, miR-34a, let-7, miR-200 and miR-15/16, are under-expressed in some cancers, including breast cancer, hepatocellular carcinoma, colon cancer, non-small-cell-lung cancer, pancreatic cancer and prostate cancer36,40. Dysfunctional expression of lncRNAs plays a vital role in the function of tumor suppressor genes and oncogenes. Approximately 15% of upregulated and 11% of downregulated lncRNAs have been reported in several types of cancer, including breast, gastric, pancreatic, lung, hepatocellular, ovarian and prostate cancer41. In a study on ovarian high-grade serous carcinoma, lncRNAs such as ADAMTS9-AS2, ACTA2-AS1, HAND2-AS1, CBR3-AS1, LINC00312, IPW, LINC00887, XIST, MEG3, TSIX and NBR2 were under-expressed42. Hyperexpression of XIST lncRNA is linked with tumor malignancies in some cancers36. The XIST may act as an oncogene, whose overexpression represses KLN2 in non-small cell lung cancers and causes miR-140/miR-124 overexpression in pancreatic carcinoma41. Overexpression of KCNQTOT1 lncRNA is implicated in colorectal cancer43. Figure 1 describes the links between epigenetic changes and cancer.
Tumor suppressor genes and cancer: Epigenetic modification of tumor suppressor genes causes loss or reduction of its function, which, combined with some factors, can lead to the cell to growing abnormally44. These genes’ loss of function may contribute more to the pathogenesis of human cancers than oncogenes’ activation45. The list of tumor suppressor genes is extensive. However, as reviewed by Joyce et al.46, the commonly encountered tumor suppressor genes include TP53, RB1, NF1, NF2, CDKN2A, WT1, BRCA1, BRCA2, PARP-1, VHL, APC, PTEN, PTCH1 and CDH1.
TP53: The TP53 tumor suppressor gene is called the “guardian of the genome” because it monitors cellular stress, such as inappropriate signaling, anoxia and DNA damage by mutated oncoproteins46,47. The TP53 gene is located on the short arm of chromosome 17 (17p13.1) and codes for the p53 protein, which regulates the expression and activity of proteins involved in apoptosis, cell cycle arrest, DNA repair and cellular senescence46. The loss of TP53 function can lead to perpetual cell replication and failure to initiate apoptosis47. The TP53 tumor suppressor is the most often mutated gene in human cancers and so the most studied gene in oncology47. It has been reported in over 50% of various forms of cancer, including colon, breast and lung cancers, leukemias, lymphomas, sarcomas and neurogenic tumors46. Hypermethylation of TP53TG1 lncRNA has been shown to lead to the loss of TP53 function in humans, resulting in cancer48. In acute myeloid leukemia, PRMT5 histone phosphorylation inhibits polo-like kinase 4 (PLK4) gene expression, activating the cGAS-STING pathway and repressing the TP53 gene49. The DNA hypermethylation has also been shown to repress the TP53 gene, resulting in hepatocellular carcinoma28.
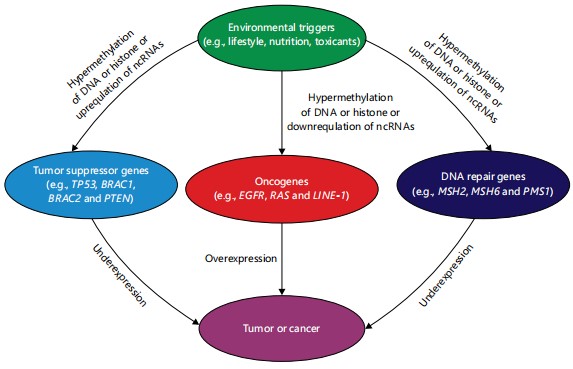
|
RB1: The RB1 gene is embedded in chromosome 13q14 and is the first identified tumor suppressor gene whose mutations predispose to cancer50. It encodes the retinoblastoma protein (pRb) and its loss of function causes retinoblastoma, breast cancer and prostate cancer50. In a study of 69 retinoblastoma patients and control groups comprising 26 normal relatives and 18 normal unrelated children, hypermethylation of the RB1 gene and DNMT genes was observed51. Overexpression of Tri-Methyl-Histone H3 Lys27 (H3K27me3) at the RB1 promoter was demonstrated to cause RB1 gene inactivation, leading to cancer52. Downregulation of let-7e, miR-320, miR-21, miR-486-3p and miR-532 has been demonstrated in the plasma of retinoblastoma patients53. The under-expression of ncRNA-RB1, a lncRNA in the promoter region of the RB1, downregulates calreticulin (CALR). The CALR is a protein in the endoplasmic reticulum that translocates to the cell surface during pre-apoptosis and serves as a signal to phagocytic cells. The ncRNA-RB1 depletion inhibits the uptake of tumor cells by macrophages54.
NF1 and NF2: The NF1 gene resides on chromosome 17q11.2 and codes for neurofibromin 1, a GTPase that negatively regulates RAS55. A germline mutation in this gene causes loss of function, resulting in neurofibromatosis type 1, an autosomal dominant disorder characterized by the development of malignant peripheral nerve sheath tumors, neurofibromas and brain tumors such as optic gliomas55. The NF2 gene is embedded in chromosome 22q12.2 and provides instructions for the production of neurofibromin 2 (otherwise called merlin), a cytoskeletal protein that is involved in contact inhibition56. Mutational loss of function in this gene leads to neurofibromatosis type 2, another autosomal dominant disorder characterized by an increased risk of tumors, particularly bilateral schwannomas56. Hypermethylation of the NF1 gene disrupts RAS/MAPK signaling, causing cutaneous neurofibromas57. Repression of NF1 in sporadic Head and Neck Squamous Cell Carcinomas (HNSCC) by overexpression of miRNA-193b activates ERK and results in tumor progression58. Decreased dimethylation and trimethylation of H3K27me3 histones at the NF1 gene cause epigenetic changes via EED or SUZ12 mutations, contributing to MPNST tumorigenesis59.
CDKN2A: This gene is situated on chromosome 9p21.3 and codes for p16/INK4a and ARF (both tumor suppressor proteins, which augment both RB and p53 functions60. Germline mutational loss of function in this gene causes autosomal dominant familial melanoma60. A biallelic mutational loss of function has been reported in several cancer types, including carcinomas, melanomas and leukemias60. Downregulation of the CDKN2A tumor suppressor gene encoding the p16INK4a protein has been reported in several cases of cancer60. The p16INK4a protein is actively involved in cell cycle regulation and senescence via its regulation of the cyclin D complexes and cyclin-dependent kinase (CDK) 4/661. In a study of colorectal cancers, CDKN2A gene promoter region was overmethylated in 61% of tumor samples62. Hypermethylation of exon 2 in CDKN2A gene in tumor, tumor-distant and tumor-adjacent tissues predisposes to breast cancer63. Abnormal histone H3K4m3 methylation and DNA methylation in the promoter region of CDKN2A gene have been detected in several cases of melanomas61. Upregulation of hsa-miR-542-5p, hsa-miR-4519 and hsa-miR-3681-3p represses CDKN2A, causing head and neck squamous cell carcinoma64.
WT1: The WT1 gene is embedded on chromosome 11 and encodes transcription factors necessary for normal genitourinary tissue development65. Germline mutational loss of function of this gene is associated with Wilms tumor, a pediatric kidney cancer65. The WT1 mutations also cause sporadic Wilms tumors65. The WT1 controls the de novo DNA methyltransferase 3A (DNMT3A) and downregulation of WT1 by short-interfering RNAs, causing the depletion of DNMT3A in human embryonal kidney-derived cell lines and Wilms tumor cells66. The DNA hypermethylation of the WT1 gene causes elevated DNMT3A, resulting in Wilms tumor cells66. The WT1 is hypermethylated and upregulated in breast cancer molecular subtypes67. The WT1 is hyperexpressed in various hematologic malignancies, particularly in myelodysplastic syndromes and acute lymphoblastic leukemia68. Hypoexpression of WT1 reduces the proliferation and increases the apoptosis of leukemic cells, indicating that WT1 might play the role of an oncogene in certain contexts68. Histone deacetylase inhibitors such as Trichostatin A (TSA) can optimally and actively repress WT1 in several cell lines68.
BRCA1, BRCA2 and PARP-1: The BRCA1 and BRCA2 are tumor suppressor genes embedded in chromosomes 17q21 and 13q12, respectively69. They code for proteins that play a role in the DNA double-strand breaks repair via the homologous recombination repair pathway69. The PARP-1 (located on 1q42.12) encodes a protein that assists in DNA single-strand breaks repair70. Lack of functional DNA repair proteins can cause the cell cycle to continue propagating defective and mutated genetic material, resulting in aberrant daughter cells. The BRCA1 gene is commonly associated with breast cancer, induced most often by the depletion or absence of its protein product. Abnormal DNA methylation is suspected as a cause of its inactivation62. Overmethylation of the gene is linked with hematologic diseases, particularly leukemia62. The BRCA1 promoter hypermethylation is higher in malignant breast tumors than in healthy adjacent tissues and benign breast lesions71. Inactivation of some miRNAs (miR-9-1, let-7, miR-29a-c, miR-148, miR-17-5p, miR-15/16, miR-27b, miR-125a/b, miR-126, miR-130a, miR-143, miR-155, miR-200c, miR-145, miR-335 and miR-205) and the overexpression of others (miR-18a, miR-10b, miR-21, miR-206 and miR-27a) cause BRCA1-related histone modifications, resulting in breast cancers72.
VHL: The Von Hippel-Lindau (VHL) gene resides on chromosome 3p25.3 and codes for a constituent of a ubiquitin ligase that plays a role in the degradation of hypoxia-induced factors (HIFs), which are transcription factors that change gene expression in response to hypoxia73. Von Hippel-Lindau syndrome is an autosomal dominant disorder, mediated by germline mutational loss of function. Inactivation of the gene poses a high risk of developing pheochromocytoma and renal cell carcinoma73. Genetic or epigenetic inactivation of the VHL, which also inactivates HIF1A, is the most suspected in renal cancer cases74. In one study, VHL promoter hypermethylation increased the risk of renal cell carcinoma75. The VHL inactivation induces global genome over methylation in human kidney cancer cells under normoxic conditions74. Histone lactylation triggered by inactive VHL contributes to clear cell renal cell carcinoma development through the activation of the transcription of platelet-derived growth factor receptor β (PDGFRβ). The PDGFRβ signaling in turn induces histone lactylation, creating an oncogenic positive feedback loop in clear-cell renal cell carcinoma75. Hyperexpression of miR-21, miR-210 and miR-155 inactivates VHL, resulting in renal cancer76.
APC: The Adenomatous polyposis coli (APC) gene is embedded in 5q22.277. This gene provides instructions for a tumor suppressor protein that regulates the WNT signaling pathway negatively, enhancing the creation of a complex that degrades β-catenin77. The β-catenin plays a role in the regulation and coordination of gene transcription and cell-cell adhesion77. The APC gene mutation is the most encountered in colon cancer and can initiate familial adenomatous polyposis (FAP)77. Loss of a normal allele of APC can cause tumor, resulting in colorectal cancer and hepatocellular carcinoma. The APC hypermethylation has been shown to cause gastrointestinal tumors, including esophageal, colorectal, gastric, pancreatic and hepatic cancer77. Overmethylation of the promoter region of APC can also predispose to breast cancer, suggesting that mutation in APC can cause other forms of cancer apart from colorectal neoplasms78. Enhanced phosphorylation levels of histones such as S2260 and T1438 in the APC gene have been observed in cancers77. The miR-494 is hyperexpressed in colorectal cancer tissues and this overexpression is negatively associated with APC expression79. The APC is targeted directly by miR-494 in colorectal cancer, with overexpression of miR-494 inducing Wnt/β-catenin signaling, promoting colorectal cancer cell progression79. Additionally, overexpression of hsa-miR-135b-5p causes loss of function of the APC gene, resulting in intestinal and diffuse gastric cancers80. The lncRNA-mAK028845 was observed to be significantly upregulated, silencing APC during colorectal cancer initiation81.
PTEN: The Phosphatase and Tensin Homolog (PTEN) gene codes for a lipid phosphatase that negatively controls the mTOR and phosphoinositide-3-kinase (PI3K)-AKT signaling pathways82. The gene is located on chromosome 10q2382. Mutations or dysregulations in PTEN can cause various cancers, including follicular thyroid cancer, breast cancer, head and neck squamous carcinoma and prostate cancer82. In a study, the PTEN methylation was significantly elevated in breast cancer blood samples than in the control and benign samples83. Additionally, hypermethylation of the PTEN gene has been shown to downregulate it by 30% in hypercoagulable blood samples compared to controls84. The PTEN relates to histone H1 and regulates chromatin condensation, suppressing tumor development and metabolism. However, overacetylation of H4K16 disrupts the association between histone H1 and PTEN, constituting regulatory feedback that may deteriorate chromatin stability85. Overexpression of miR-130a decreases PTEN levels, activating the PI3K/Akt/eNOS (endothelial nitric oxide synthase) signaling pathway, enhancing injury and inflammatory responses in Human Coronary Artery Endothelial Cells (HCAECs)86. In a study, overexpression of miR-301a directly targeted and suppressed PTEN, promoting breast cancer87. The miR-21 post-transcriptionally downregulates PTEN expression to promote cell proliferation and cervical cancer cell survival88. In laryngeal squamous cell carcinoma, lncRNA HOTAIR interacts with miR-29b and represses PTEN89.
PTCH1: The PTCH1 tumor suppressor gene resides on chromosome 9q22.3 and encodes the protein patched homolog 1, which negatively regulates the hedgehog signaling pathway90. Germline mutational loss of function of this gene predisposes to Gorlin syndrome, an autosomal dominant disorder that poses a high risk of developing medulloblastoma and basal cell carcinoma90. Acquired biallelic loss of function of PTCH1 mutations is often linked with sporadic cases of medulloblastoma and basal cell carcinoma90. Overmethylation of the promoter region of the PTCH1 gene is the main cause of reduced PTCH1 expression in gastric cancer AGS cells91. Hypermethylation of the CpG islands of the PTCH1 gene was observed in AGS gastric cancer cells, suggesting that overmethylation of the promoter region of the PTCH1 gene is involved in the carcinogenesis and development of gastric cancer92. The miR-9 is overexpressed in Glioblastoma Multiforme (GBM) patients and causes resistance to temozolomide, a drug used for treating glioblastoma that targets the Sonic Hedgehog receptor PTCH193. This suggests that overexpression of miR-9 downregulates PTCH1. In a study, miR-9 expression was raised in the tissues of GBM patients and early-passage GBM cell lines of patients with recurrent GBM but not from naïve patients93. Protein phosphorylation and autophosphorylation of the PTCH1 gene, such as protein kinase C phosphorylation and peptidyl-tyrosine, are implicated in poor prognosis and increased recurrence of breast cancer94. Histone Deacetylase (HDAC) modification has been shown to decrease PTCH1 gene expression, causing breast cancer95.
CDH1: The CDH1 (E-cadherin) gene is a tumor suppressor gene that resides on chromosome 16q22.196. Once in contact with neighboring cells, healthy cells stop replicating, which maintains the structure and architecture of the tissue, a phenomenon called contact inhibition96. The E-cadherin binds to β-catenin, an important constituent of the WNT signaling pathway, to regulate contact inhibition96. Germline mutational loss of function in this gene is associated with autosomal dominant familial gastric carcinoma96. Loss of function mutation and hypermethylation of the CDH1 gene were observed in breast cancer patients in Kashmir97. The CDH1 gene promoter hypermethylation has been documented in gastric cancer and chronic gastritis98. Underacetylation of histones H4K16Ac and H3 in the exon 8 of the CDH1 gene has been reported in gastric cancer cell lines99. Additionally, histone methylation analysis showed elevated H3K36 trimethylation in the exon 8 of the CDH1 gene in gastric cancer cell lines compared to healthy cells99. In a study, repression of hsa-miR-383 in association with upregulation of AL356608.1, LINC00337, AL357153.2, MALAT1, HULC and LINC00485 interacted with CDH1 to promote breast cancer100. The miR-383 functionally suppresses tumor, inhibiting cell replication, metastasis and epithelial-mesenchymal transition (EMT) in breast cancer by targeting ETS129. Furthermore, miR-205, miR-200a and miR-429 are often hyper-expressed in pancreatic tumors, while CDH1 is under-expressed101. Long non-coding RNA H19 mediates methylation-dependent repression of the CDH1 promoter, contributing to the progression of lung adenocarcinoma102.
Oncogenes and cancer: The oncogenes are numerous, but some are frequently implicated in cancer. Several proto-oncogenes are functionally involved in embryogenesis and specifically stimulate cell proliferation and growth during organismal development103. Additionally, some negatively regulate cell differentiation103. Typically, proto-oncogenes are switched off after completing the developmental processes they regulate103. However, if proto-oncogene remains active or is faultily reactivated as the individuals get older, it may induce cancer103. Some frequently implicated oncogenes include Ras, EGFR, EML4AK, LINE-1 and SAT2.
Ras gene family: The Ras genes produce proteins that regulate how cells receive signals, differentiate, grow and die. The Ras gene family-KRAS, NRAS and HRAS-are the most suspected oncogenes in human tumors104. The three genes accounted for about 20-30% of all human malignancies, including 25% of lung carcinomas and about 50% of colon carcinomas104. Mutational activation of the KRAS gene is suspected in about 25% of various forms of cancers105,106. The KRAS mutations are responsible for almost 85% of all RAS mutations in human tumors, with NRAS accounting for between 11 and 15% and HRAS for about 1%106. The KRAS G12V hyperexpression in an isogenic lung model shows more than 50,600 demethylated CpGs compared to non-transformed controls107. In a study, overexpression of KRAS induced upregulation of miR-30c and miR-21, resulting in lung cancer108. In another study, overexpression of HRAS in MKN-28 cells by siRNA increased the replication, angiogenesis and metastasis of gastric carcinoma cells, while repressing endogenous HRAS induced the opposite109. Overexpression of NRAS boosts tumor development by stimulating the secretion of IL8 through JAK2 activation and has been reported to be overexpressed in basal-like breast cancer110. Overexpression of miRNA-708 has been shown to successfully reduce the levels of NRAS protein in lung cancer, leukemia and melanoma cell lines with NRAS mutations, suppressing anchorage-independent growth, formation of reactive oxygen species-induced apoptosis and cell proliferation104. In a study, specific CpG sites demethylation in the first intron of R-RAS activated over half of gastric cancers111. When siRNA was introduced into the R-RAS-expressing cells, the adhered cells disappeared, which suggests that functional blocking of the R-RAS-signaling pathway could be an effective therapeutic strategy for gastric cancer111.
EGFR and EML4-ALK genes: The Epidermal Growth Factor Receptor (EGFR) gene is a member of the HER/ERB-B family of transmembrane receptor kinases and resides on chromosome 7p11.2112. The EGFR protein is involved in cell signaling pathways that regulate cell division and survival112. Certain types of cancer cells produce EGFR proteins in higher-than-normal amounts due to mutations in the EGFR gene112. Hyperexpression of EGFR decreases cell proliferation, survival and migration in multiple solid tumors113. However, in a study, hypermethylation (90%) and a moderate degree of methylation (30-50%) of EGFR were observed in breast cancer cell lines113. Abnormal histone H3K9 and H3K27 methylation promotes EGFR amplification and has been reported in several cases of pancreatic cancer114. The miR-34a downregulation has been shown to cause overexpression of EGFR, leading to the initiation of several forms of cancers, including non-small cell lung cancer115. In a study involving breast cancer cell lines, miR-218 upregulation downregulated EGFR, leading to the downregulation of p44/42 MAPK signaling116. In another study involving human non-small cell lung cancer cell lines, lncRNA LINC00240 served as a sponge for miR-7-5p and caused upregulation of EGFR117.
LINE-1: Hypomethylation of long-interspersed nuclear element-1 (LINE-1) gene (locus: 22 q12.1) is linked with cell proliferation and malignant traits in lung adenocarcinoma118. The LINE-1 are "jumping genes" often called L1 retrotransposons and constitute 17% of human DNA. They multiply throughout the genome using a “copy-and-paste” mechanism mediated by RNA, a process termed retrotransposition119. The L1s are switched on in the gamete cells and are actively involved in embryogenesis, but are turned off in somatic cells via epigenetic mechanisms119. However, in cancer cells, L1s are wrongly activated and may induce genome instability, a hallmark of carcinogenesis119. Hypomethylation of LINE-1 is suspected in several cancer types and is linked with a worse prognosis. In a study of 1,211 colorectal cancer patients, LINE-1 hypomethylation was observed120. In another study, hypermethylation of LINE-1 DNA was observed in the white blood cells of pancreatic cancer patients121. Downregulation of let-7 miRNA has been shown to lead to overexpression of LINE-1 in lung cancer patients122.
SAT2: Spermidine/spermine N1-acetyl transferase 2(SAT2) gene, a locus mapping within the 1q12 pericentromeric region, codes for an enzyme known as diamine acetyltransferase 2123. This enzyme regulates an important metabolic glutamine/glutamate balance underpinning retrograde signaling by dendritic release of the neurotransmitter glutamate124. Loss of function SAT2 gene has been reported to cause cancer. Notably, DNA hypomethylation of the SAT2 gene accelerates tumor progression in ovarian cancer and early onset of tumor in breast cancer125. In a study involving several cancer cell lines, upregulation of the SAT2 gene by heat shock demethylation was observed126. In another study, hypomethylation of SAT2 DNA was detected in individuals with ovarian cancer127.
DNA repair genes and cancer: Exogenous and endogenous DNA-damaging agents, including ionizing radiation, chemotherapeutic agents and ultraviolet light can cause DNA lesions128. These lesions include single-strand breaks, mismatches, double-strand breaks, interstrand or intrastrand cross-links and chemical modifications of the bases or sugars128. Cells encounter more than 20,000 DNA-damaging events daily129. To counteract this damage, cells initiate DNA repair processes that tolerate or remove the damage, contributing to genomic integrity. The repair process takes a variable amount of time, depending on the substrate130,131. Thus, making DNA repair the first defensive strategy against genotoxic stress, albeit without lasting effects129,132. Further to its cancer prevention role, DNA repair impacts cancer treatment outcomes with DNA-damaging drugs and radiation129. The efficiency of DNA repair is mainly governed by the repair gene’s expression levels, which can be altered by several factors. These factors include mutations in their promoter or coding regions, expression levels of transcription factors, and/or epigenetic factors such as CpG promoter methylation or demethylation and histone modifications129. Thus, defects in DNA repair genes are a common cause of cancer132.
Over 200 DNA repair proteins have been documented and their roles in DNA repair have been outlined129. Five main DNA repair pathways are active throughout different stages of the cell cycle, enabling cells to repair DNA damage. These pathways include nucleotide excision repair (NER), base excision repair (BER), mismatch repair (MMR), DNA crosslink repair (Fanconi anemia genes), homologous recombination (HR) and non-homologous end joining (NHEJ)130. The BER pathway repairs single-strand breaks, while the NHEJ and HR pathways repair double-strand breaks132. The MMR pathway fixes single nucleotide mismatches, deletion and aberrant nucleotide insertions132. The NER pathway repairs helix-distorting lesions such as ultraviolet radiation-induced pyrimidine dimers132. The Fanconi anemia (FA) pathway, involving downstream repair nucleases, a core complex and ubiquitin ligase recognizes and repairs interstrand crosslinks. The direct reversal (DR) pathway utilizes O6-methylguanine DNA methyltransferase to remove damaging DNA methylations132.
Several epigenetic aberrations in DNA repair genes have been implicated in cancer. Notably, silencing mutations and hypermethylation in MMR genes (e.g., MLH1, MSH2, PMS2, MSH6 and PMS1) result in microsatellite instability and Lynch syndrome, increasing the risk of cancer, particularly colorectal cancer132. Germline mutational loss of BRCA1 and BRCA2 involved in FA and HR repair has been reported to increase the risk of ovarian and breast cancer132. Aberrations in the ATM and HR genes are suspected in ataxia telangiectasia and malignancy risk, while NER abnormalities in xeroderma pigmentosum are linked with an increased risk of skin cancer132.
EPIGENETICS IN THE DIAGNOSIS AND MANAGEMENT OF CANCER
Epigenetic factors interact with genetic and non-genetic factors to influence biological events in the body, including disease development. To this end, epigenetic applications such as epigenetic biomarkers, epigenetic drugs and epigenetic editing have been developed to diagnose and treat diseases in the clinic133,134. Notably, cancer cells exhibit increased DNA methylation at certain genes but have overall lower DNA methylation levels compared to normal cells135. This difference allows for the use of epigenetics to differentiate cancerous cells from healthy ones. Additionally, different types of cancer that appear similar can have distinct DNA methylation patterns; thus, epigenetics can help determine the specific type of cancer and detect hard-to-identify cancers earlier135. Furthermore, the expression patterns of non-coding RNAs (ncRNAs) are strongly tissue-specific, particularly in cancer. These ncRNAs can be released from cancer cells into blood or urine, serving as diagnostic markers or prognostic indicators28,36.
Currently, DNA methylation of specific genes or the relative expression of microRNAs are the most common epigenetic-based in vitro diagnostic tests. Both are often carried out using pyrosequencing technologies and RT-qPCR-based methods (e.g., methylation-sensitive high-resolution melting, methyl-specific PCR and methyLight)136. Commercially available epigenetic tests include the EPICUP® assay, the EpiproColon® test (Epigenomics AG, Berlin, Germany), Beadchip 450K (Illumina) and Cologuard® Stool DNA-Based Test (Exact Sciences Corp., Madison, WI), among others137. Epigenetic biomarkers aid in disease progression monitoring, early diagnosis, patient selection and stratification by risk, disease outcome prediction, evaluation of therapeutic interventions in specific patient subsets and prediction of future comorbidities137. Furthermore, microRNAs and DNA methylation are very stable compared to proteins and RNA, making these epigenetic biomarkers more viable and practical in clinical procedures137. Epigenetic biomarkers are dynamic in nature, they provide information about gene function and specific genetic programs altered during disease development, all of which make them preferable over protein-based or genetic biomarkers137. This suggests that epigenetics has great potential to improve predictive and precision medicine137. However, epigenetics alone cannot diagnose cancer; additional screening tests are needed to confirm cancer135.
Some epigenetic drugs are currently in clinical trials, while others have been tested and found safe and effective138. Numerous small-molecule inhibitors that target chromatin- and histone-modifying enzymes to reverse abnormal epigenetic changes in tumors have passed clinical trials as cancer therapeutics. These include Azacitidine, Nanaomycin A, Belinostat and Tubacin, among others22. Pharmacological inhibitors of DNA methylation, such as azacitidine and decitabine, have also been developed to reverse epigenetic changes via DNA methylation inhibition22. Anti-miRNA oligonucleotides, otherwise called Anti-miRs, are commercially available and include Eteplirsen (Exondys 51) and Golodirsen (Vyondys 53 TM)139,140. However, some of these drugs exhibit poor pharmacokinetic and safety/tolerability issues due to a lack of specificity and off-target effects141. Additionally, their therapeutic outcomes varied in solid tumors due to the lack of biomarker-driven targeted therapies, resulting in a highly heterogeneous response22. Recently, significant progress has been made in this regard, with the formulation of epigenomic therapies that directly address these issues by targeting defined loci with highly precise, durable and tunable approaches141.
CONCLUSION
The findings of this review demonstrate that epigenetic changes, such as DNA methylation, histone post-translational modifications and noncoding RNA silencing, are important for biological processes, particularly growth and development. These changes are influenced by lifestyle, environmental factors and nutritional status, which can lead to aberrant epigenetic reconfigurations and result in diseases. Specifically, cancer is most often mediated by abnormal epigenetic alterations in tumor suppressor genes (e.g., TP53, RB1, NF1, NF2, CDKN2A, WT1, BRCA1, BRCA2, PARP-1, VHL, APC, PTEN, PTCH1 and CDH1), oncogenes (e.g., Ras, EGFR, EML4AK, LINE-1 and SAT2), or DNA repair genes (e.g., MSH2, MSH6, MLH1, PMS1 and PMS2). Epigenetic changes are reversible, making drugs that target these changes potentially more effective in predisposed individuals. Some drugs, known as epigenetic drugs, such as Azacitidine, Belinostat and Tubacin, have shown promise but face challenges related to pharmacokinetics and safety. However, these issues are currently being addressed. More studies are needed to improve the precision of these drugs and ensure that non-target genes or epigenomes are not affected. Additionally, because epigenetic therapeutic procedures are complex, further research is required to simplify them.
SIGNIFICANCE STATEMENT
Epigenetic reprogramming plays a critical role in the increasing prevalence of diseases, particularly cancers, yet its significance in disease development remains underrecognized. This synthesis of existing literature underscores the links between epigenetic modifications and cancer etiology, highlighting their potential as therapeutic targets. Abnormal epigenetic changes in tumor suppressor genes, oncogenes and DNA repair genes are strongly associated with cancer development. Notably, the reversibility of some epigenetic aberrations using approved drugs, such as Azacitidine, Belinostat and Tubacin, underscores the promise of epigenetic therapies. Clinical trials reveal that these therapies improve outcomes in specific cancer patient populations, suggesting their integration into standard treatment regimens could yield significant benefits. However, challenges such as poor drug specificity and off-target effects highlight the need for further research to improve the precision and safety of epigenetic interventions, advancing the development of targeted and effective cancer therapies.
REFERENCES
- Farsetti, A., B. Illi and C. Gaetano, 2023. How epigenetics impacts on human diseases. Eur. J. Intern. Med., 114: 15-22.
- Mazzone, R., C. Zwergel, M. Artico, S. Taurone, M. Ralli, A. Greco and A. Mai, 2019. The emerging role of epigenetics in human autoimmune disorders. Clin. Epigenet., 11.
- Deichmann, U., 2016. Epigenetics: The origins and evolution of a fashionable topic. Dev. Biol., 416: 249-254.
- Ospelt, C., 2022. A brief history of epigenetics. Immunol. Lett., 249: 1-4.
- Lacal, I. and R. Ventura, 2018. Epigenetic inheritance: Concepts, mechanisms and perspectives. Front. Mol. Neurosci., 11.
- Loscalzo, J. and D.E. Handy, 2014. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease (2013 Grover Conference Series). Pulm. Circ., 4: 169-174.
- Moosavi, A. and A.M. Ardekani, 2016. Role of epigenetics in biology and human diseases. Iran. Biomed. J., 20: 246-258.
- Al Aboud, N.M., C. Tupper and I. Jialal, 2023. Genetics, Epigenetic Mechanism. StatPearls Publishing, Treasure Island.
- Oppermann, U., 2013. Why is epigenetics important in understanding the pathogenesis of inflammatory musculoskeletal diseases? Arthritis Res. Ther., 15.
- Montgomery, M. and A. Srinivasan, 2019. Epigenetic gene regulation by dietary compounds in cancer prevention. Adv. Nutr., 10: 1012-1028.
- Komers, R., D. Mar, O. Denisenko, B. Xu, T.T. Oyama and K. Bomsztyk, 2013. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab. Invest., 93: 543-552.
- Kowluru, R.A., 2020. Retinopathy in a diet-induced type 2 diabetic rat model and role of epigenetic modifications. Diabetes, 69: 689-698.
- Hamdane, N., F. Jühling, E. Crouchet, H. El Saghire and C. Thumann et al., 2019. HCV-induced epigenetic changes associated with liver cancer risk persist after sustained virologic response. Gastroenterology, 156: 2313-2329.E7.
- Sallam, M., M. Mysara, M.A. Benotmane, R. Tamarat and S.C.R. Santos et al., 2022. DNA methylation alterations in fractionally irradiated rats and breast cancer patients receiving radiotherapy. Int. J. Mol. Sci., 23.
- Yahaya, T.O., D.M. Bashar, E.O. Oladele, J. Umar, D. Anyebe and A. Izuafa, 2022. Epigenetics in the etiology and management of infertility. World J. Med. Genet., 10: 7-21.
- Yahaya, T., E. Oladele, A. Muhammed, A. Haruna and U. Liman, 2021. Involvement of epigenetics in the pathogenesis, testing and management of Coronavirus disease 2019 (COVID-19) pandemic: A narrative review. Pharm. Biomed. Res., 7: 161-170.
- Fatiregun, O.A., O. Bakare, S. Ayeni, A. Oyerinde and A.C. Sowunmi et al., 2020. 10-year mortality pattern among cancer patients in Lagos State University Teaching Hospital, Ikeja, Lagos. Front. Oncol., 10. .
- Bray, F., M. Laversanne, H. Sung, J. Ferlay, R.L. Siegel, I. Soerjomataram and A. Jemal, 2024. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J. Clinicians, 74: 229-263.
- Brown, J.S., S.R. Amend, R.H. Austin, R.A. Gatenby, E.U. Hammarlund and K.J. Pienta, 2023. Updating the definition of cancer. Mol. Cancer Res., 21: 1142-1147.
- Debela, D.T., S.G.Y. Muzazu, K.D. Heraro, M.T. Ndalama and B.W. Mesele et al., 2021. New approaches and procedures for cancer treatment: Current perspectives. SAGE Open Med., 9.
- Yuzhalin, A.E., 2024. Redefining cancer research for therapeutic breakthroughs. Br. J. Cancer, 130: 1078-1082.
- Yu, X., H. Zhao, R. Wang, Y. Chen and X. Ouyang et al., 2024. Cancer epigenetics: From laboratory studies and clinical trials to precision medicine. Cell Death Discovery, 10.
- Lai, J., Z. Liu, Y. Zhao, C. Ma and H. Huang, 2021. Anticancer effects of I-BET151, an inhibitor of bromodomain and extra-terminal domain proteins. Front. Oncol., 11.
- Liang, T., F. Wang, R.M. Elhassan, Y. Cheng and X. Tang et al., 2023. Targeting histone deacetylases for cancer therapy: Trends and challenges. Acta Pharm. Sin. B, 13: 2425-2463.
- Bure, I.V., M.V. Nemtsova and E.B. Kuznetsova, 2022. Histone modifications and non-coding RNAs: Mutual epigenetic regulation and role in pathogenesis. Int. J. Mol. Sci., 23.
- Wu, Y.L., Z.J. Lin, C.C. Li, X. Lin and S.K. Shan et al., 2023. Epigenetic regulation in metabolic diseases: Mechanisms and advances in clinical study. Signal Transduction Targeted Ther., 8.
- Zhao, Z. and A. Shilatifard, 2019. Epigenetic modifications of histones in cancer. Genome Biol., 20.
- Sun, Y.M. and Y.Q. Chen, 2020. Principles and innovative technologies for decrypting noncoding RNAs: From discovery and functional prediction to clinical application. J. Hematol. Oncol., 13.
- Zhang, P., W. Wu, Q. Chen and M. Chen, 2019. Non-coding RNAs and their integrated networks. J. Integr. Bioinf., 16.
- Pathak, A., S. Tomar and S. Pathak, 2023. Epigenetics and cancer: A comprehensive review. Asian Pac. J. Cancer Biol., 8: 75-89.
- Gu, M., B. Ren, Y. Fang, J. Ren and X. Liu et al., 2024. Epigenetic regulation in cancer. MedComm, 5.
- Tang, Q., J. Cheng, X. Cao, H. Surowy and B. Burwinkel, 2016. Blood-based DNA methylation as biomarker for breast cancer: A systematic review. Clin. Epigenet., 8.
- Chen, C., Z. Wang, Y. Ding, L. Wang, S. Wang, H. Wang and Y. Qin, 2022. DNA methylation: From cancer biology to clinical perspectives. Front. Bioscience-Landmark, 27.
- Boycott, C., M. Beetch, T. Yang, K. Lubecka and Y. Ma et al., 2022. Epigenetic aberrations of gene expression in a rat model of hepatocellular carcinoma. Epigenetics, 17: 1513-1534.
- López-Jiménez, E. and E. Andrés-León, 2021. The implications of ncRNAs in the development of human diseases. Non-Coding RNA, 7.
- Yan, H. and P. Bu, 2021. Non-coding RNA in cancer. Essays Biochem., 65: 625-639.
- Hu, M.H., C.Y. Ma, X.M. Wang, C.D. Ye, G.X. Zhang, L. Chen and J.G. Wang, 2016. MicroRNA-126 inhibits tumor proliferation and angiogenesis of hepatocellular carcinoma by down-regulating EGFL7 expression. Oncotarget, 7: 66922-66934.
- Kim, T. and C.M. Croce, 2023. MicroRNA: Trends in clinical trials of cancer diagnosis and therapy strategies. Exp. Mol. Med., 55: 1314-1321.
- Ullmann, P., M. Nurmik, M. Schmitz, F. Rodriguez and J. Weiler et al., 2019. Tumor suppressor miR-215 counteracts hypoxia-induced colon cancer stem cell activity. Cancer Lett., 450: 32-41.
- Otmani, K. and P. Lewalle, 2021. Tumor suppressor miRNA in cancer cells and the tumor microenvironment: Mechanism of deregulation and clinical implications. Front. Oncol., 11.
- Guzel, E., T.M. Okyay, B. Yalçınkaya, S. Karacaoglu, M. Gocmen and M.H. Akçakuyu, 2020. Tumor suppressor and oncogenic role of long non-coding RNAs in cancer. Northern Clin. İstanbul, 7: 81-86.
- Hayashi-Okada, M., S. Sato, K. Nakashima, T. Sakai and T. Tamehisa et al., 2024. Identification of long noncoding RNAs downregulated specifically in ovarian high-grade serous carcinoma. Reprod. Med. Biol., 23.
- Liu, J., W. Lv, S. Li and J. Deng, 2021. Regulation of long non-coding RNA KCNQ1OT1 network in colorectal cancer immunity. Front. Genet., 12.
- Velez, A.M.A. and M.S. Howard, 2015. Tumor-suppressor genes, cell cycle regulatory checkpoints, and the skin. N. Am. J. Med. Sci., 7: 176-188.
- Weinberg, R.A., 2014. The Biology of Cancer. 2nd Edn., Garland Science, New York, ISBN: 9780815342199, Pages: 876.
- Joyce, C., A. Rayi and A. Kasi, 2023. Tumor-Suppressor Genes. StatPearls Publishing, Treasure Island.
- Wang, H., M. Guo, H. Wei and Y. Chen, 2023. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduction Targeted Ther., 8.
- Diaz-Lagares, A., A.B. Crujeiras, P. Lopez-Serra, M. Soler and F. Setien et al., 2016. Epigenetic inactivation of the p53-induced long noncoding RNA TP53 target 1 in human cancer. Proc. Natl. Acad. Sci. U.S.A., 113: E7535-E7544.
- Man, C.H., W. Lam, C.C. Dang, X.Y. Zeng and L.C. Zheng et al., 2023. Inhibition of PLK4 remodels histone methylation and activates the immune response via the cGAS-STING pathway in TP53-mutated AML. Blood, 142: 2002-2015.
- Berry, J.L., A. Polski, W.K. Cavenee, T.P. Dryja, A.L. Murphree and B.L. Gallie, 2019. The RB1 story: Characterization and cloning of the first tumor suppressor gene. Genes, 10.
- Yazici, H., H.C. Wu, H. Tigli, E.Z. Yilmaz, R. Kebudi and R.M. Santella, 2020. High levels of global genome methylation in patients with retinoblastoma. Oncol. Lett., 20: 715-723.
- Wen, X., T. Ding, F. Li, J. Fan, X. Fan, R. Jia and H. Zhang, 2022. Interruption of aberrant chromatin looping is required for regenerating RB1 function and suppressing tumorigenesis. Commun. Biol., 5.
- Liu, S.S., Y.S. Wang, Y.F. Sun, L.X. Miao and J. Wang et al., 2014. Plasma microRNA-320, microRNA-let-7e and microRNA-21 as novel potential biomarkers for the detection of retinoblastoma. Biomed. Rep., 2: 424-428.
- Musahl, A.S., X. Huang, S. Rusakiewicz, E. Ntini and A. Marsico et al., 2015. A long non-coding RNA links calreticulin-mediated immunogenic cell removal to RB1 transcription. Oncogene, 34: 5046-5054.
- Anastasaki, C., P. Orozco and D.H. Gutmann, 2022. RAS and beyond: The many faces of the neurofibromatosis type 1 protein. Dis. Models Mech., 15.
- Yuan, R., B. Wang, Y. Wang and P. Liu, 2024. Gene therapy for neurofibromatosis type 2-related schwannomatosis: Recent progress, challenges, and future directions. Oncol. Ther., 12: 257-276.
- Grit, J.L., B.K. Johnson, P.S. Dischinger, C.J. Essenburg and M. Adams et al., 2021. Distinctive epigenomic alterations in NF1-deficient cutaneous and plexiform neurofibromas drive differential MKK/p38 signaling. Epigenet. Chromatin, 14.
- Lenarduzzi, M., A.B.Y. Hui, N.M. Alajez, W. Shi and J. Williams et al., 2013. MicroRNA-193b enhances tumor progression via down regulation of neurofibromin 1. PLoS ONE, 8.
- Wang, W., C.J. Wei, X.W. Cui, Y.H. Li and Y.H. Gu et al., 2021. Impacts of NF1 gene mutations and genetic modifiers in neurofibromatosis type 1. Front. Neurol., 12.
- Chan, S.H., J. Chiang and J. Ngeow, 2021. CDKN2A germline alterations and the relevance of genotype-phenotype associations in cancer predisposition. Hereditary Cancer Clin. Pract., 19.
- Zhao, R., B.Y. Choi, M.H. Lee, A.M. Bode and Z. Dong, 2016. Implications of genetic and epigenetic alterations of CDKN2A (p16 INK4a) in cancer. EBioMedicine, 8: 30-39.
- Das, P.M. and R. Singal, 2004. DNA methylation and cancer. J. Clin. Oncol., 22: 4632-4642.
- Spitzwieser, M., E. Entfellner, B. Werner, W. Pulverer, G. Pfeiler, S. Hacker and M. Cichna-Markl, 2017. Hypermethylation of CDKN2A exon 2 in tumor, tumor-adjacent and tumor-distant tissues from breast cancer patients. BMC Cancer, 17.
- Gopalakrishnan, S., A. Pandi, P. Arumugam and V.P. Jayaseelan, 2024. MicroRNAs targeting CDKN2A gene as a potential prognostic marker in head and neck squamous cell carcinoma. Mol. Biol. Res. Commun., 13: 21-27.
- Torban, E. and P. Goodyer, 2024. Wilms’ tumor gene 1: Lessons from the interface between kidney development and cancer. Am. J. Physiol. Renal Physiol., 326: F3-F19.
- Szemes, M., A.R. Dallosso, Z. Melegh, T. Curry and Y. Li et al., 2013. Control of epigenetic states by WT1 via regulation of de novo DNA methyltransferase 3A. Hum. Mol. Genet., 22: 74-83.
- Ren, C., X. Tang and H. Lan, 2021. Comprehensive analysis based on DNA methylation and RNA-seq reveals hypermethylation of the up-regulated WT1 gene with potential mechanisms in PAM50 subtypes of breast cancer. PeerJ, 9.
- Shahidul Makki, M., T. Heinzel and C. Englert, 2008. TSA downregulates Wilms tumor gene 1 (Wt1) expression at multiple levels. Nucleic Acids Res., 36: 4067-4078.
- Stoppa-Lyonnet, D., 2016. The biological effects and clinical implications of BRCA mutations: Where do we go from here? Eur. J. Hum. Genet., 24: S3-S9.
- Pandey, N. and B.E. Black, 2021. Rapid detection and signaling of DNA damage by PARP-1. Trends Biochem. Sci., 46: 744-757.
- Oubaddou, Y., M. Oukabli, S. Fenniche, A. Elktaibi and M.R. Elochi et al., 2023. BRCA1 promoter hypermethylation in malignant breast tumors and in the histologically normal adjacent tissues to the tumors: Exploring its potential as a biomarker and its clinical significance in a translational approach. Genes, 14.
- Downs, B. and S.M. Wang, 2015. Epigenetic changes in BRCA1-mutated familial breast cancer. Cancer Genet., 208: 237-240.
- Ohh, M., C.C. Taber, F.G. Ferens and D. Tarade, 2022. Hypoxia-inducible factor underlies von Hippel-Lindau disease stigmata. eLife, 11.
- Artemov, A.V., S. Zhenilo, D. Kaplun, A. Starshin and A. Sokolov et al., 2022. An IDH-independent mechanism of DNA hypermethylation upon VHL inactivation in cancer. Epigenetics, 17: 894-905.
- Yang, J., L. Luo, C. Zhao, X. Li and Z. Wang et al., 2022. A positive feedback loop between inactive VHL-triggered histone lactylation and PDGFRβ signaling drives clear cell renal cell carcinoma progression. Int. J. Biol. Sci., 18: 3470-3483.
- Neal, C.S., M.Z. Michael, L.H. Rawlings, M.B. van der Hoek and J.M. Gleadle, 2010. The VHL-dependent regulation of microRNAs in renal cancer. BMC Med., 8.
- Zhang, Y., X. Liu, A. Li and X. Tang, 2022. A pan-cancer analysis on the carcinogenic effect of human adenomatous polyposis coli. PLoS ONE, 17.
- Zhu, L., X. Li, Y. Yuan, C. Dong and M. Yang, 2021. APC promoter methylation in gastrointestinal cancer. Front. Oncol., 11.
- Zhang, Y., L. Guo, Y. Li, G.H. Feng, F. Teng, W. Li and Q. Zhou, 2018. MicroRNA-494 promotes cancer progression and targets adenomatous polyposis coli in colorectal cancer. Mol. Cancer, 17.
- Magalhães, L., L.G. Quintana, D.C.F. Lopes, A.F. Vidal and A.L. Pereira et al., 2018. APC gene is modulated by hsa-miR-135b-5p in both diffuse and intestinal gastric cancer subtypes. BMC Cancer, 18.
- Li, J., Y. Ji, N. Chen, H. Wang and C. Fang et al., 2022. A specific upregulated long noncoding RNA in colorectal cancer promotes cancer progression. JCI Insight, 7.
- He, Y., M.M. Sun, G.G. Zhang, J. Yang, K.S. Chen, W.W. Xu and B. Li, 2021. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduction Targeted Ther., 6.
- Ramadan, A., M. Hashim, A. Abouzid and M. Swellam, 2021. Clinical impact of PTEN methylation status as a prognostic marker for breast cancer. J. Genet. Eng. Biotechnol., 19.
- Hoseini, M., M. Sahmani, F. Foroughi, Y.K. Monfared and M. Azad, 2019. Evaluating the role of PTEN promoter methylation in patients predisposed to hypercoagulable states via methylation specific PCR. Rep. Biochem. Mol. Biol., 7: 223-229.
- Chen, Z.H., M. Zhu, J. Yang, H. Liang and J. He et al., 2014. PTEN interacts with histone H1 and controls chromatin condensation. Cell Rep., 8: 2003-2014.
- Song, C.L., B. Liu, Y.F. Shi, N. Liu and Y.Y. Yan et al., 2016. MicroRNA-130a alleviates human coronary artery endothelial cell injury and inflammatory responses by targeting PTEN via activating PI3K/Akt/eNOS signaling pathway. Oncotarget, 7: 71922-71936.
- Ma, F., J. Zhang, L. Zhong, L. Wang and Y. Liu et al., 2014. Upregulated microRNA-301a in breast cancer promotes tumor metastasis by targeting PTEN and activating Wnt/β-catenin signaling. Gene, 535: 191-197.
- Peralta-Zaragoza, O., J. Deas, A. Meneses-Acosta, F. de la O-Gómez and G. Fernández-Tilapa et al., 2016. Relevance of miR-21 in regulation of tumor suppressor gene PTEN in human cervical cancer cells. BMC Cancer, 16.
- Li, D., J. Feng, T. Wu, Y. Wang, Y. Sun, J. Ren and M. Liu, 2013. Long intergenic noncoding RNA HOTAIR is overexpressed and regulates PTEN methylation in laryngeal squamous cell carcinoma. Am. J. Pathol., 182: 64-70.
- Ponti, G., A. Pollio, L. Pastorino, G. Pellacani and C. Magnoni et al., 2012. Patched homolog 1 gene mutation (p.G1093R) induces nevoid basal cell carcinoma syndrome and non-syndromic keratocystic odontogenic tumors: A case report. Oncol. Lett., 4: 241-244.
- Zuo, Y., Y. Song, M. Zhang, Z. Xu and X. Qian, 2014. Role of PTCH1 gene methylation in gastric carcinogenesis. Oncol. Lett., 8: 679-682.
- Zuo, Y. and Y. Song, 2013. Detection and analysis of the methylation status of PTCH1 gene involved in the hedgehog signaling pathway in a human gastric cancer cell line. Exp. Ther. Med., 6: 1365-1368.
- Munoz, J.L., V. Rodriguez-Cruz, S.H. Ramkissoon, K.L. Ligon, S.J. Greco and P. Rameshwar, 2015. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget, 6: 1190-1201.
- Wang, C.Y., Y.C. Chang, Y.L. Kuo, K.T. Lee and P.S. Chen et al., 2019. Mutation of the PTCH1 gene predicts recurrence of breast cancer. Sci. Rep., 9.
- Ecke, I., F. Petry, A. Rosenberger, S. Tauber, S. Mönkemeyer and I. Hess et al., 2009. Antitumor effects of a combined 5-Aza-2′deoxycytidine and valproic acid treatment on rhabdomyosarcoma and medulloblastoma in Ptch mutant mice. Cancer Res., 69: 887-895.
- Shenoy, S., 2019. CDH1 (E-Cadherin) mutation and gastric cancer: Genetics, molecular mechanisms and guidelines for management. Cancer Manage. Res., 11: 10477-10486.
- Asiaf, A., S.T. Ahmad, S.A. Aziz, A.A. Malik, Z. Rasool, A. Masood and M.A. Zargar, 2014. Loss of expression and aberrant methylation of the CDH1 (E-cadherin) gene in breast cancer patients from Kashmir. Asian Pac. J. Cancer Prev., 15: 6397-6403.
- Bayat, M., A. Shirgir, A.K. Veisari and R.N. Sadeghi, 2024. Detection of CDH1 gene promoter hypermethylation in gastric cancer and chronic gastritis. Pract. Lab. Med., 40.
- Li, X.W., B.Y. Shi, Q.L. Yang, J. Wu and H.M. Wu et al., 2015. Epigenetic regulation of CDH1 exon 8 alternative splicing in gastric cancer. BMC Cancer, 15.
- Fan, T., L. Xue, B. Dong, H. He and W. Zhang et al., 2022. CDH1 overexpression predicts bladder cancer from early stage and inversely correlates with immune infiltration. BMC Urol., 22.
- Diaz-Riascos, Z.V., M.M. Ginesta, J. Fabregat, T. Serrano and J. Busquets et al., 2019. Expression and role of microRNAs from the miR-200 family in the tumor formation and metastatic propensity of pancreatic cancer. Mol. Ther. Nucleic Acids, 17: 491-503.
- Gao, L.M., S.F. Xu, Y. Zheng, P. Wang and L. Zhang et al., 2019. Long non-coding RNA H19 is responsible for the progression of lung adenocarcinoma by mediating methylation-dependent repression of CDH1 promoter. J. Cell. Mol. Med., 23: 6411-6428.
- Dakal, T.C., B. Dhabhai, A. Pant, K. Moar and K. Chaudhary et al., 2024. Oncogenes and tumor suppressor genes: Functions and roles in cancers. MedComm, 5.
- Pang, J.M., P.C. Chien, M.C. Kao, P.Y. Chiu and P.X. Chen et al., 2023. MicroRNA-708 emerges as a potential candidate to target undruggable NRAS. PLoS ONE, 18.
- Hobbs, G.A., C.J. Der and K.L. Rossman, 2016. RAS isoforms and mutations in cancer at a glance. J. Cell Sci., 129: 1287-1292.
- Kodaz, H., O. Kostek, M.B. Hacioglu, B. Erdogan and C.E. Kodaz et al., 2017. Frequency of RAS mutations (KRAS, NRAS, HRAS) in human solid cancer. Eurasian J. Med. Oncol., 1: 1-7.
- Tew, B.Y., J.K. Durand, K.L. Bryant, T.K. Hayes and S. Peng et al., 2020. Genome-wide DNA methylation analysis of KRAS mutant cell lines. Sci. Rep., 10.
- Shi, L., J. Middleton, Y.J. Jeon, P. Magee and D. Veneziano et al., 2018. KRAS induces lung tumorigenesis through microRNAs modulation. Cell Death Dis., 9.
- Wu, X.Y., W.T. Liu, Z.F. Wu, C. Chen and J.Y. Liu et al., 2016. Identification of HRAS as cancer-promoting gene in gastric carcinoma cell aggressiveness. Am. J. Cancer Res., 6: 1935-1948.
- Zheng, Z.Y., L. Tian, W. Bu, C. Fan and X. Gao et al., 2015. Wild-type N-Ras, overexpressed in basal-like breast cancer, promotes tumor formation by inducing IL-8 secretion via JAK2 activation. Cell Rep., 12: 511-524.
- Nishigaki, M., K. Aoyagi, I. Danjoh, M. Fukaya and K. Yanagihara et al., 2005. Discovery of aberrant expression of R-RAS by cancer-linked DNA hypomethylation in gastric cancer using microarrays. Cancer Res., 65: 2115-2124.
- Roskoski Jr. R., 2014. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res., 79: 34-74.
- Montero, A.J., C.M. Díaz-Montero, L. Mao, E. Youssef, M.R. Estecio, L. Shen and J.P. Issa, 2006. Epigenetic inactivation of EGFR by CpG island hypermethylation in cancer. Cancer Biol. Ther., 5: 1494-1501.
- Clarke, T.L., R. Tang, D. Chakraborty, C. van Rechem and F. Ji et al., 2020. Histone lysine methylation dynamics control EGFR DNA copy-number amplification. Cancer Discovery, 10: 306-325.
- Li, Y.L., X.M. Liu, C.Y. Zhang, J.B. Zhou and Y. Shao et al., 2017. MicroRNA-34a/EGFR axis plays pivotal roles in lung tumorigenesis. Oncogenesis, 6.
- Wischmann, F.J., F.M. Troschel, M. Frankenberg, B. Kemper and A.V. Kumar et al., 2023. Tumor suppressor miR-218 directly targets epidermal growth factor receptor (EGFR) expression in triple-negative breast cancer, sensitizing cells to irradiation. J. Cancer Res. Clin. Oncol., 149: 8455-8465.
- Ku, G.W., Y. Kang, S.L. Yu, J. Park and S. Park et al., 2021. LncRNA LINC00240 suppresses invasion and migration in non-small cell lung cancer by sponging miR-7-5p. BMC Cancer, 21.
- Kitahara, H., T. Okamoto, S. Shimamatsu, M. Kohno and Y. Morodomi et al., 2020. LINE-1 hypomethylation is associated with malignant traits and cell proliferation in lung adenocarcinoma. Anticancer Res., 40: 5659-5666.
- Xiao-Jie, L., X. Hui-Ying, X. Qi, X. Jiang and M. Shi-Jie, 2016. LINE-1 in cancer: Multifaceted functions and potential clinical implications. Genet. Med., 18: 431-439.
- Inamura, K., M. Yamauchi, R. Nishihara, P. Lochhead and Z.R. Qian et al., 2014. Tumor LINE-1 methylation level and microsatellite instability in relation to colorectal cancer prognosis. JNCI: J. Nat. Cancer Inst., 106.
- Neale, R.E., P.J. Clark, J. Fawcett, L. Fritschi and B.N. Nagler et al., 2014. Association between hypermethylation of DNA repetitive elements in white blood cell DNA and pancreatic cancer. Cancer Epidemiol., 38: 576-582.
- Tristán-Ramos, P., A. Rubio-Roldan, G. Peris, L. Sánchez and S. Amador-Cubero et al., 2020. The tumor suppressor microRNA let-7 inhibits human LINE-1 retrotransposition. Nat. Commun., 11.
- Baek, J.H., Y.V. Liu, K.R. McDonald, J.B. Wesley, M.E. Hubbi, H. Byun and G.L. Semenza, 2007. Spermidine/spermine-N1-acetyltransferase 2 is an essential component of the ubiquitin ligase complex that regulates hypoxia-inducible factor 1α. J. Biol. Chem., 282: 23572-23580.
- Jenstad, M., A.Z. Quazi, M. Zilberter, C. Haglerød and P. Berghuis et al., 2009. System A transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cereb. Cortex, 19: 1092-1106.
- Dor, Y. and H. Cedar, 2018. Principles of DNA methylation and their implications for biology and medicine. Lancet, 392: 777-786.
- Tilman, G., N. Arnoult, S. Lenglez, A. van Beneden, A. Loriot, C. de Smet and A. Decottignies, 2012. Cancer-linked satellite 2 DNA hypomethylation does not regulate Sat2 non-coding RNA expression and is initiated by heat shock pathway activation. Epigenetics, 7: 903-913.
- Widschwendter, M., G. Jiang, C. Woods, H.M. Müller and H. Fiegl et al., 2004. DNA hypomethylation and ovarian cancer biology. Cancer Res., 64: 4472-4480.
- Li, L.Y., Y.D. Guan, X.S. Chen, J.M. Yang and Y. Cheng, 2021. DNA repair pathways in cancer therapy and resistance. Front. Pharmacol., 11.
- Christmann, M. and B. Kaina, 2019. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. Rev. Mutat. Res., 780: 15-28.
- Chatterjee, N. and G.C. Walker, 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen., 58: 235-263.
- Huang, R. and P.K. Zhou, 2021. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduction Targeted Ther., 6.
- Chae, Y.K., J.F. Anker, B.A. Carneiro, S. Chandra and J. Kaplan et al., 2016. Genomic landscape of DNA repair genes in cancer. Oncotarget, 7: 23312-23321.
- Yahaya, T., E. Oladele, U. Shemishere and M. Abdulrau’f, 2019. Role of epigenetics in the pathogenesis and management of type 2 diabetes mellitus. Transl: Univ. Toledo J. Med. Sci., 6: 20-28.
- Sarvari, P., P. Sarvari, I. Ramírez-Díaz, F. Mahjoubi and K. Rubio, 2022. Advances of epigenetic biomarkers and epigenome editing for early diagnosis in breast cancer. Int. J. Mol. Sci., 23.
- Albulescu, A., A. Plesa, A. Fudulu, I.V. Iancu, G. Anton and A. Botezatu, 2021. Epigenetic approaches for cervical neoplasia screening (Review). Exp. Ther. Med., 22.
- García-Giménez, J.L., S. Mena-Mollá, J. Beltrán-García and F. Sanchis-Gomar, 2017. Challenges in the analysis of epigenetic biomarkers in clinical samples. Clin. Chem. Lab. Med., 55: 1474-1477.
- Beltrán-García, J., R. Osca-Verdegal, S. Mena-Mollá and J.L. García-Giménez, 2019. Epigenetic IVD tests for personalized precision medicine in cancer. Front. Genet., 10.
- Tajudeen, Y.O. and U.B. Shemishere, 2019. Role of epigenetics in aetiology and therapies for Type 1 diabetes mellitus: A narrative review. J. Health Social Sci., 4: 199-212.
- Taufiqul Arif, K.M., E.K. Elliott, L.M. Haupt and L.R. Griffiths, 2020. Regulatory mechanisms of epigenetic miRNA relationships in human cancer and potential as therapeutic targets. Cancers, 12.
- Menon, A., N. Abd-Aziz, K. Khalid, C.L. Poh and R. Naidu, 2022. miRNA: A promising therapeutic target in cancer. Int. J. Mol. Sci., 23.
- Feehley, T., C.W. O’Donnell, J. Mendlein, M. Karande and T. McCauley, 2023. Drugging the epigenome in the age of precision medicine. Clin. Epigenet., 15.
How to Cite this paper?
APA-7 Style
Tajudeen,
Y., John,
E., Hewage,
A.S., Liman,
U.U., Caleb,
O. (2025). Epigenetic Modifications in Cancer Etiology, Diagnosis and Therapy. Asian Journal of Biological Sciences, 18(2), 516-532. https://doi.org/10.3923/ajbs.2025.516.532
ACS Style
Tajudeen,
Y.; John,
E.; Hewage,
A.S.; Liman,
U.U.; Caleb,
O. Epigenetic Modifications in Cancer Etiology, Diagnosis and Therapy. Asian J. Biol. Sci 2025, 18, 516-532. https://doi.org/10.3923/ajbs.2025.516.532
AMA Style
Tajudeen
Y, John
E, Hewage
AS, Liman
UU, Caleb
O. Epigenetic Modifications in Cancer Etiology, Diagnosis and Therapy. Asian Journal of Biological Sciences. 2025; 18(2): 516-532. https://doi.org/10.3923/ajbs.2025.516.532
Chicago/Turabian Style
Tajudeen, Yahaya, Emmanuel John, Asanka Sudeshini Hewage, Umar Usman Liman, and Omede Caleb.
2025. "Epigenetic Modifications in Cancer Etiology, Diagnosis and Therapy" Asian Journal of Biological Sciences 18, no. 2: 516-532. https://doi.org/10.3923/ajbs.2025.516.532

This work is licensed under a Creative Commons Attribution 4.0 International License.